
Nueva herramienta contra la tuberculosis
Investigadores argentinos descubrieron una serie de compuestos que podría ser el inicio de un desarrollo farmacológico efectivo en el tratamiento de la tuberculosis. Mediante técnicas de bioinformática, combinadas con experimentos, identificaron con precisión una proteína clave de la enfermedad y detectaron las características de los compuestos necesarias para inhibirla.
La tuberculosis es una enfermedad causada por Mycobacterium tuberculosis, una bacteria que casi siempre afecta a los pulmones. Es prevenible y curable, pero si no se trata oportunamente puede causar daño permanente. Es la decimotercera causa de muerte y la enfermedad infecciosa más mortífera por detrás del COVID-19. Se calcula que una cuarta parte de la población mundial tiene tuberculosis latente, es decir, que están infectadas por el bacilo pero aún no han enfermado ni pueden transmitir la infección. Pero aun así, debido a factores ambientales o de salud, la bacteria puede salir de la latencia y producir una infección aguda.
Según datos de la OMS, por primera vez en más de una década, en 2020 aumentó la mortalidad por tuberculosis (1,5 millones de muertes). Además, la inversión mundial en prevención, diagnóstico y tratamiento de la tuberculosis en ese año no llegó ni a la mitad de la meta mundial prevista para 2022 (13.000 millones de dólares anuales), más allá de que acabar con la epidemia de tuberculosis es una de las metas de los Objetivos de Desarrollo Sostenible (ODS) relacionados con la salud. En lo referente a Argentina, según cifras del Ministerio de Salud de la Nación, durante 2020 se notificaron en nuestro país 10.896 casos de tuberculosis, con 656 muertes, y la tasa nacional se ubica en 24,01 casos por cada 100 mil habitantes.
Se trata de una enfermedad infecciosa desatendida (EID), propia de los países en vías de desarrollo, que se agrava ante el costo de producción de nuevos fármacos o bien por el desinterés de las compañías farmacéuticas en producirlos. A partir del descenso sostenido en la inversión de fármacos contra la tuberculosis (ver reporte de la OMS sobre esta problemática), especialmente en la producción de antibióticos resistentes a nuevas cepas, surge la pregunta sobre cómo mejorar el desarrollo de estos fármacos para que sean más efectivos y menos costosos, e innovar en su producción.
¿Qué hace que una proteína del patógeno de la tuberculosis sea atractiva para el desarrollo de un fármaco?
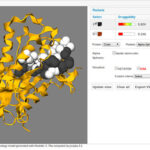
Ante este complejo escenario, un grupo de investigadores en bioinformática de la Facultad de Ciencias Exactas y Naturales de la UBA se encuentra trabajando desde hace una década en el estudio del genoma de la bacteria que causa la tuberculosis. Tomando la información genética y molecular de la patología, se realiza el análisis de las interacciones de todos los fármacos conocidos, de modo de poder encontrar las mejores características de una droga más efectiva contra la enfermedad. Tan es así que el paradigma de descubrimiento de nuevas drogas, utilizando bioinformática, consiste en encontrar una molécula capaz de unirse e inhibir o activar eficientemente un blanco macromolecular específico, generalmente una proteína. Este proceso da lugar a poder identificar, diseñar y optimizar nuevos fármacos, que sean específicos y potentes.
La pregunta que, en este caso, buscan responder los científicos sería: ¿qué hace que una proteína del patógeno de la tuberculosis sea atractiva desde el punto de vista del desarrollo de un fármaco, como para que pueda ser utilizada como blanco molecular, es decir, una proteína clave que si se pudiera modular su actividad podría tener un efecto terapéutico?

Adrián Turjanski. Foto: Archivo Exactas UBA
«Desde un principio nosotros diseñamos una base de datos con toda la información que hoy conocemos sobre el genoma de la tuberculosis (y de otras enfermedades), luego integramos los datos en un servidor que se llama Target Pathogen y buscamos allí los genes que nos parecían potenciales blancos moleculares. Nuestros algoritmos computacionales determinaron que una de las proteínas, quinasa G, resulta muy influyente en el metabolismo de la tuberculosis» puntualiza Adrián Turjanski, investigador de CONICET y director del Laboratorio de Bioinformática Estructural de Exactas UBA.
“Nuestros algoritmos computacionales determinaron que una de las proteínas, quinasa G, resulta muy influyente en el metabolismo de la tuberculosis”.
Turjanski, también investigador del Instituto de Química Biológica (IQUIBICEN), aclara que esa proteína en particular presenta propiedades claves para su estudio. En este sentido, la enfermedad tiene un estado de latencia (queda adentro del organismo esperando que nuestras defensas bajen hasta infectarnos) y cuando la tuberculosis se manifiesta produce la activación de las células del sistema inmunológico denominadas «macrófagos» (un tipo de leucocitos o glóbulos blancos), capaces de fagocitar diversas partículas y destruir microorganismos. De este modo, se desencadena la respuesta inmune cuya finalidad es atacar a los agentes infecciosos de la enfermedad y neutralizar su patogenicidad o manifestación de sus síntomas. Los macrófagos fagocitan al bacilo pero este tiene la capacidad de persistir dentro del cuerpo en un estado de latencia. La quinasa G, mencionada anteriormente, es capaz de regular el metabolismo de la tuberculosis para entrar en este estado. Por lo tanto, esta proteína cumple un rol importantísimo en el organismo y es susceptible de ser inhibida por drogas que ayuden al sistema inmune a matar a la tuberculosis.
«Al tener claro este panorama, tomamos la estructura química de esa proteína e hicimos experimentos para identificar qué lugar de la proteína era el mejor para inhibir y qué características debían tener las moléculas para lograr ese resultado. Esto nos permitió filtrar 20 mil drogas de todas las posibles. Nuestra metodología, que llevó años desarrollar, utiliza simulaciones donde se observa la proteína entera en agua, sus movimientos, en qué lugar el disolvente se queda adherido y, a partir de las interacciones relevantes que identificamos, pudimos calcular con precisión qué tipo de droga, qué interacciones debía tener con la proteína y en qué posición debía estar», explica Turjanski. Y prosigue comentando el arduo proceso que llevó al hallazgo de los compuestos para los nuevos fármacos: “Conociendo qué propiedades tenía que tener la droga, consultamos nuevamente la base de datos de los 20 mil compuestos, los filtramos por nuestro tamiz de software y calculamos tanto la estructura de la interacción como la energía de interacción, ya que esa energía es proporcional a cuánto inhibe. Este cálculo nos permitió quedarnos con solo cuarenta drogas de las 20 mil. Luego, pudimos comprar y en algunos casos sintetizar esas drogas, y realizar ensayos de inhibición tanto in-vitro como in-vivo. De esta manera, obtuvimos tres nuevas drogas capaces de ejercer el efecto esperado: demostraron ser las mejores inhibidoras en macrófagos infectados», afirma. La metodología, que se puede aplicar a cualquier patógeno, permite encontrar eficientemente nuevos inhibidores para después realizar con mayor éxito todos los ensayos clínicos, disminuyendo así los altos costos que se requieren.
“Obtuvimos tres nuevas drogas capaces de ejercer el efecto esperado: demostraron ser las mejores inhibidoras en macrófagos infectados”.
El artículo con los resultados de estos métodos de descubrimiento de fármacos fue recientemente publicado en Journal of Medicinal Chemistry. “El universo de blancos moleculares es prácticamente infinito y las técnicas de bioinformática estructural orientadas al desarrollo de medicamentos ayudan a predecir qué moléculas pequeñas son capaces de unirse a qué proteínas. Este descubrimiento tiene una relevancia particular para la comunidad científica, médica y farmacéutica. Por un lado encontramos qué características debe tener un compuesto en general para inhibir una proteína y tenemos una herramienta que identifica muy bien blancos moleculares, ya que otros investigadores podrían querer atacar otro gen de la tuberculosis. Por otro, considerando que nuestra base de datos de proteínas y compuestos es pública, otros investigadores podrían utilizarla para el estudio de más enfermedades y sus posibles fármacos”, sostiene Marcelo Marti, Investigador de CONICET en el instituto IQUIBICEN e integrante del proyecto.

Marcelo Martí. Foto: Exactas UBA.
Marti amplía las diversas posibilidades de uso de la base de datos del Laboratorio de Bioinformática Estructural: “Tenemos una base de datos de enfermedades desatendidas. Esta base nuclea toda la información relacionada con los blancos moleculares y los fármacos de parásitos y bacterias afines a microorganismos que producen enfermedades que afectan fundamentalmente a los países en vías de desarrollo. No solo tuberculosis, sino también dengue, chagas y leishmaniasis, entre otras”. El científico comenta que esta información les ha posibilitado mantener estrechas colaboraciones con investigadores de Brasil, Uruguay y otros países de la región, con el fin de llevar a cabo investigaciones que permitan optimizar el desarrollo de fármacos y disminuir el costo que conlleva su producción.
Un cambio de paradigma en el desarrollo de medicamentos
En las últimas décadas, la bioinformática ha contribuido significativamente al boom de la biología molecular mediante el desarrollo de algoritmos para el análisis, clasificación y secuenciación de ADN y proteínas. Pero la bioinformática estructural no se ha quedado atrás: mediante el conocimiento de características fisicoquímicas de los componentes de las biomacromoléculas, se han desarrollado algoritmos que permiten generar modelos de estructuras a partir de información obtenida por técnica de rayos X y resonancia magnética nuclear. En el comienzo del siglo XXI, el extraordinario avance en la secuencia de genomas transformó profundamente el panorama de la biología, y abrió un abanico de oportunidades para el uso de estructuras tridimensionales en la búsqueda de fármacos.

Dario Fernandez Do Porto.
Acompañando este proceso, desde el Laboratorio de Bioinformática Estructural, junto a investigadores del IQUIBICEN y del Instituto de Cálculo, se han podido desarrollar bases de datos donde se encuentran disponibles la totalidad de los genomas de distintos patógenos. “Nuestra plataforma web Target Pathogen permite integrar los datos de patógenos relevantes desde el punto de vista clínico. La ventaja es que los datos se pueden «rankear», de acuerdo al interés de cada investigador en particular o la enfermedad que desee estudiar, para priorizar una pequeña lista de aquellas proteínas con atractivos desde el punto de vista de los blancos moleculares. Y todo ello, tiene una validación experimental: una vez que determinamos que un blanco sería esencial para el crecimiento bacterial, esto nos permite hacer foco en las proteínas que resultan más atractivas”, comenta Dario Fernandez Do Porto, Investigador en Ciencias Biológicas del Instituto de Cálculo, integrante del proyecto y uno de los principales responsables del desarrollo de Target Pathogen.
Por su parte, Do Porto comenta que en este momento disponen de treinta patógenos con relevancia clínica pero que el desarrollo está abierto a cualquier grupo o investigador que esté trabajando actualmente con un patógeno que no esté presente en la plataforma y le interese subir su genoma, “pueden recurrir a la plataforma y capacitarse en su uso aplicado. De hecho hubo grupos que, de ese modo, empezaron colaboraciones que luego han resultado en publicaciones muy valiosas. Y si el genoma ya está en nuestra plataforma, pueden usarla porque es totalmente abierta y muy amigable para el usuario, incluso la utilizan estudiantes”, concluye. Y recalca que esta tecnología permite abaratar los costos en los procesos usuales para el desarrollo de un fármaco, reduciendo la cantidad de ensayos clínicos, así como también maximiza las probabilidades de éxito en la tarea de poder determinar con precisión el blanco molecular de cada compuesto.
Claramente la bioinformática no consiste sólo en el estudio algorítmico o el análisis de bases de datos de secuencias y de proteínas. La comparación de secuencias o búsquedas en bases de datos son sólo la punta del iceberg: cuando la bioinformática es combinada con la creciente información estructural y el conocimiento de las bases físico-químicas que gobiernan el comportamiento de los sistemas biológicos a nivel atómico molecular, se convierte en una herramienta de proyecciones inimaginables para continuar con el estudio de nuevas enfermedades y el desarrollo de mejores fármacos para combatirlas.