
Computadora mata tubo de ensayo
Algunos investigadores han reemplazado la mesada del laboratorio por la pantalla de la computadora y, por lo tanto, algunos experimentos tradicionales se concretan a través de sofisticados programas de computación. La experimentación simulada arroja un detalle nunca visto a la hora de entender y trabajar sobre las interacciones químicas

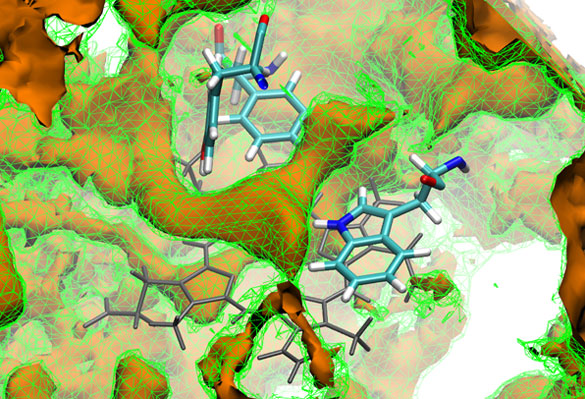
Simulación de hemoglobina truncada de Thermobifida fusca. GMM
Quienes tuvimos la suerte de ir a la escuela llevamos en algún lugar de la memoria el recuerdo de los trabajos prácticos que realizábamos en el laboratorio del colegio. Aquellas primeras experiencias “científicas” –que nos amontonaban alrededor de un microscopio para tratar de identificar una célula, o que nos compelían a mezclar sustancias para ver un cambio de color– daban cuenta de un universo extraño, poblado de un montón de instrumentos raros.
Que el mundo cambia y nada es como era antes es una verdad de Perogrullo. También es una perogrullada afirmar que el desarrollo de la informática ha cambiado nuestras vidas de una manera radical. Pero lo que –hasta el momento– escapa del ámbito de lo obvio es que las computadoras están reemplazando a los tubos de ensayo, las probetas y las pipetas que usábamos en aquellos experimentos escolares.
Es así. La realidad virtual ya no es solamente patrimonio de los juegos de computación. Ahora, los propios científicos hacen experimentos virtuales: “Así como un simulador de vuelo es capaz de determinar si un piloto es capaz de llevar el avión a destino, la química computacional puede determinar, por ejemplo, si cierta droga es capaz de inhibir a una enzima o si un par de proteínas pueden interactuar entre sí”, ilustra el doctor Adrián Turjanski, investigador del Conicet en el Instituto de Química Física de los Materiales, Medioambiente y Energía (INQUIMAE) y en el Departamento de Química Inorgánica, Analítica, y Química Física (DQIAyQF) dela Facultadde Ciencias Exactas y Naturales (FCEyN) dela UBA.
¿Por qué utilizar computadoras en lugar de tubos de ensayo? Por ejemplo, para entender qué sucede durante una reacción química. Porque al efectuar un experimento real en un tubo podremos saber que, si mezclamos una sustancia A con otra sustancia B, ellas podrán reaccionar entre sí para dar un producto C. Pero de esta manera es casi imposible conocer al detalle lo que ha ocurrido en el intermedio.
Así, las computadoras ayudan a llenar ese bache en el conocimiento, a entender en profundidad cómo se comportan los átomos durante una reacción química. O también, por ejemplo, a predecir qué conducta seguirán dos moléculas en el momento en que se encuentren.
Juego de química
Para jugar con los átomos y las moléculas en la computadora, los científicos deben recurrir a programas de computación diseñados para tal fin: “La química computacional se vale de modelos matemáticos. Resolviendo las ecuaciones correspondientes, estos modelos permiten entender las interacciones entre átomos y moléculas y predecir comportamientos de la materia que en ocasiones es difícil estudiar en el laboratorio”, explica el doctor Darío Estrin, investigador del Conicet en el DQIA y QF/INQUIMAE y director del Grupo de Modelado Molecular.
Dichos modelos matemáticos son conjuntos de ecuaciones diseñadas a partir del conocimiento teórico que se tiene acerca de cómo se comporta la materia en diferentes condiciones y son los que permiten “dar vida” a los átomos y a las moléculas y “verlos” moverse e interaccionar en la pantalla de la computadora.
En términos generales, existen dos tipos de modelos para efectuar simulaciones: “Una manera de simular el comportamiento de las moléculas es a través de los llamados métodos clásicos, que son modelos más sencillos basados en las leyes de la física clásica. Pero los más precisos son los que están basados en las leyes de la mecánica cuántica y que permiten describir los fenómenos al nivel de los electrones”, señala Estrin. Cabe aclarar que la mecánica cuántica es una rama de la física que estudia el comportamiento de la materia y cuyas leyes rigen el mundo de lo muy pequeño.
Los límites del juego
Algunas reacciones químicas suelen ocurrir muy rápido. Muchísimo más rápido que un abrir y cerrar de ojos. “En términos generales pueden durar nanosegundos –o sea, milmillonésimas de segundos–, y algunas son todavía más rápidas: pueden ocurrir en picosegundos –billonésimas de segundos– o, incluso, en menos tiempo”, ejemplifica Estrin.
Por lo tanto, para estudiar una reacción química podría ser suficiente con simular unos pocos microsegundos, es decir, unos pocos miles de nanosegundos de la vida de las moléculas.
Parece muy poco. Sin embargo, lo efímero de este fenómeno no implica que simular in silico (en la computadora) lo que ocurre in vivo sea sencillo. De hecho, tiene un alto costo de procesamiento y, por lo tanto, límites. Porque las ecuaciones con las que se construyen los modelos son complejas.
“Si bien son más precisos, la principal desventaja de los modelos basados en las leyes de la mecánica cuántica es la complejidad de sus ecuaciones, por lo que los cálculos requieren mucho en recursos de computación y, por lo tanto, en tiempo de procesamiento. En cambio, los modelos basados en la física clásica demandan menos recursos de computación, pero solo permiten estudiar fenómenos que no incluyan ruptura o formación de enlaces químicos”, comenta Estrin.
Así como la capacidad de procesamiento establece un límite temporal para la simulación, es decir, para cuánto tiempo de vida de la molécula se puede estudiar, la potencia de computación disponible también limita el tamaño del sistema a analizar, porque cuanto mayor sea la molécula en estudio, mayor será la necesidad de recursos de procesamiento.
“Si tengo un sistema muy chiquito, podría predecir su evolución durante más tiempo que si tuviera un sistema más grande”, consigna Estrin, y da un ejemplo: “Con los modelos clásicos, yo puedo simular sistemas más grandes durante más tiempo. Por ejemplo, puedo simular un microsegundo de una proteína de 200 aminoácidos, que igual es muy poquito. Por otro lado, con un modelo cuántico, el límite es mucho más chiquito todavía. Por ejemplo, su aplicación se limita a moléculas formadas por no más de cien átomos”.
En definitiva, la simulación por computadora de la vida de las moléculas está limitada por el poder de cómputo. Para sobrellevar esta restricción tecnológica y obtener alto poder de cálculo, los investigadores recurren a los denominados “clusters”, es decir, a un conjunto de computadoras interconectadas en serie. Por ejemplo, en la FCEyN existe el Centro de Cómputos de Alto Rendimiento (CeCAR) que dispone de 56 computadoras interconectadas, cada una de las cuales tiene cuatro microprocesadores, lo que resulta en que el cluster tiene un total de 224 procesadores trabajando simultáneamente.
Para dar una idea de la potencia del cluster del CeCAR (en el mundo existen otros más potentes), Turjanski da un ejemplo: “Estudiar diez nanosegundos de la vida de una proteína con una computadora, analizar con una sola máquina qué le sucede a esa proteína picosegundo a picosegundo durante diez nanosegundos puede llevar meses, y con el cluster eso se puede hacer en pocos días”.
Pero todavía hay fronteras que aun el cluster más poderoso es incapaz de atravesar: “Si quiero estudiar cómo una droga entra y sale de una proteína, es un fenómeno que ocurre en el orden de los milisegundos, y nadie lo puede simular hoy en día, ni siquiera con las computadoras más potentes que existen”, señala.
Para Estrin, estas dificultades pueden aliviarse en cierta medida utilizando criterios muy personales a la hora de determinar el sistema de estudio: “Si bien dependemos de la capacidad de la computadora, la intuición química del investigador puede ayudar de alguna manera. Por ejemplo, cuando uno está observando un fenómeno reactivo, sabemos que este no ocurre en toda la molécula sino en una parte de ese sistema y, por lo tanto, limitamos la simulación a ese lugar del sistema”.
Juego y realidad
Se puede jugar con las moléculas en una pantalla para poder explicar comportamientos observados experimentalmente, o para predecir esos comportamientos cuando el experimento es complicado o irrealizable. También, se pueden hacer simulaciones para ver qué modificaciones se le podrían hacer a un determinado medicamento para que sea más activo, sin necesidad de estar probando alternativas engorrosas en el laboratorio. De igual manera, se puede simular químicamente un compuesto para encontrar métodos alternativos para su producción.
“La química computacional produjo un cambio de paradigma en el desarrollo de medicamentos, contribuyendo al diseño racional de drogas y acelerando los procesos de identificación y mejoramiento de nuevos compuestos”, informa Turjanski.
La simulación del comportamiento de átomos y moléculas en la computadora presenta, además, ventajas potenciales en otros campos, como la nanotecnología y la electrónica molecular.
No obstante, todavía, más que imitar fielmente el comportamiento de la naturaleza lo que se han logrado son aproximaciones bastante cercanas a la realidad: “Todas estas simulaciones se basan en algún modelo y, por lo tanto, nunca van a ser mejores que lo que el modelo permita. No es lo mismo que un experimento real, en el sentido de que en el experimento real puede haber problemas de interpretación, pero el resultado es unívoco. Acá también hay un resultado unívoco, pero para el modelo que uno está simulando, que no necesariamente va a coincidir con la realidad”, aclara Estrin.
En cualquier caso, los resultados alcanzados hasta el momento han demostrado que la química computacional es una disciplina útil para generar conocimiento nuevo.
Quizás, algún día sea posible calcar de manera fiel la realidad y que asistamos a la extinción de los tubos, las probetas, las pipetas y toda la batería de artefactos que componen un laboratorio real. Quizás, algún día, los experimentos escolares se realicen en grandes salas repletas de computadoras.



